Research

HiMSELF: A Hierarchical Misbehavior Classification with Sequence Embedding by Latent Features in Vehicular Ad-Hoc Networks
Vehicular ad hoc network (VANET) enables vehicles, infrastructure, and pedestrians to exchange information, enhancing safety, efficiency, and intelligent decision-making. VANETs operate by exchanging basic safety messages (BSMs) between vehicles and infrastructure. However, as network connectivity increases, so does the risk of malfunctions or malicious data injections that can cause severe disruptions. Accordingly, a misbehavior detection system (MDS) to detect and analyze abnormal data patterns and potential attack behaviors has become essential. In particular, identifying misbehavior classification based on specific abnormal types is essential for establishing a robust and comprehensive VANET security framework. To address these challenges, a hierarchical misbehavior classification method with sequence embedding by latent features, referred to as HiMSELF, is proposed. HiMSELF, defined as a hierarchical classification system (HCS), employs a trained deep learning (DL) model to classify various misbehavior types, embed their intrinsic representations, and perform hierarchical clustering. The resulting structure reflects the intrinsic relationships among the types and serves as the basis for constructing the HCS. The HiMSELF classification pipeline operates in two sequential stages. First, the system employsBSMsequence data as input and grouped into broader, higher-level categories associated with each sample. Subsequently, it identifies the specific misbehavior type within the corresponding category. In experiments classifying 19 misbehavior types, HiMSELF achieved an average F1-score of 0.9918, outperforming existing approaches and demonstrating its potential to underpin reliable security mechanisms in cooperative intelligent transportation systems (C-ITS).

Controllable Mask Diffusion Model for medical annotation synthesis with semantic information extraction
Medical segmentation, a prominent task in medical image analysis utilizing artificial intelligence, plays a crucial role in computer-aided diagnosis and depends heavily on the quality of the training data. However, the availability of sufficient data is constrained by strict privacy regulations associated with medical data. To mitigate this issue, research on data augmentation has gained significant attention. Medical segmentation tasks require paired datasets consisting of medical images and annotation images, also known as mask images, which represent lesion areas or radiological information within the medical images. Consequently, it is essential to apply data augmentation to both image types. This study proposes a Controllable Mask Diffusion Model, a novel approach capable of controlling and generating new masks. This model leverages the binary structure of the mask to extract semantic information, namely, the mask’s size, location, and count, which is then applied as multi-conditional input to a diffusion model via a regressor. Through the regressor, newly generated masks conform to the input semantic information, thereby enabling input-driven controllable generation. Additionally, a technique that analyzes correlation within semantic information was devised for large-scale data synthesis. ...

ReCDAP: Relation-Based Conditional Diffusion with Attention Pooling for Few-Shot Knowledge Graph Completion
Knowledge Graphs (KGs), composed of triples in the form of (head, relation, tail) and consisting of entities and relations, play a key role in information retrieval systems such as question answering, entity search, and recommendation. In real-world KGs, although many entities exist, the relations exhibit a long-tail distribution, which can hinder information retrieval performance. Previous few-shot knowledge graph completion studies focused exclusively on the positive triple information that exists in the graph or, when negative triples were incorporated, used them merely as a signal to indicate incorrect triples. To overcome this limitation, we propose Relation-Based Conditional Diffusion with Attention Pooling (ReCDAP). First, negative triples are generated by randomly replacing the tail entity in the support set. By conditionally incorporating positive information in the KG and non-existent negative information into the diffusion process, the model separately estimates the latent distributions for positive and negative relations. Moreover, including an attention pooler enables the model to leverage the differences between positive and negative cases explicitly. Experiments on two widely used datasets demonstrate that our method outperforms existing approaches, achieving state-of-the-art performance.
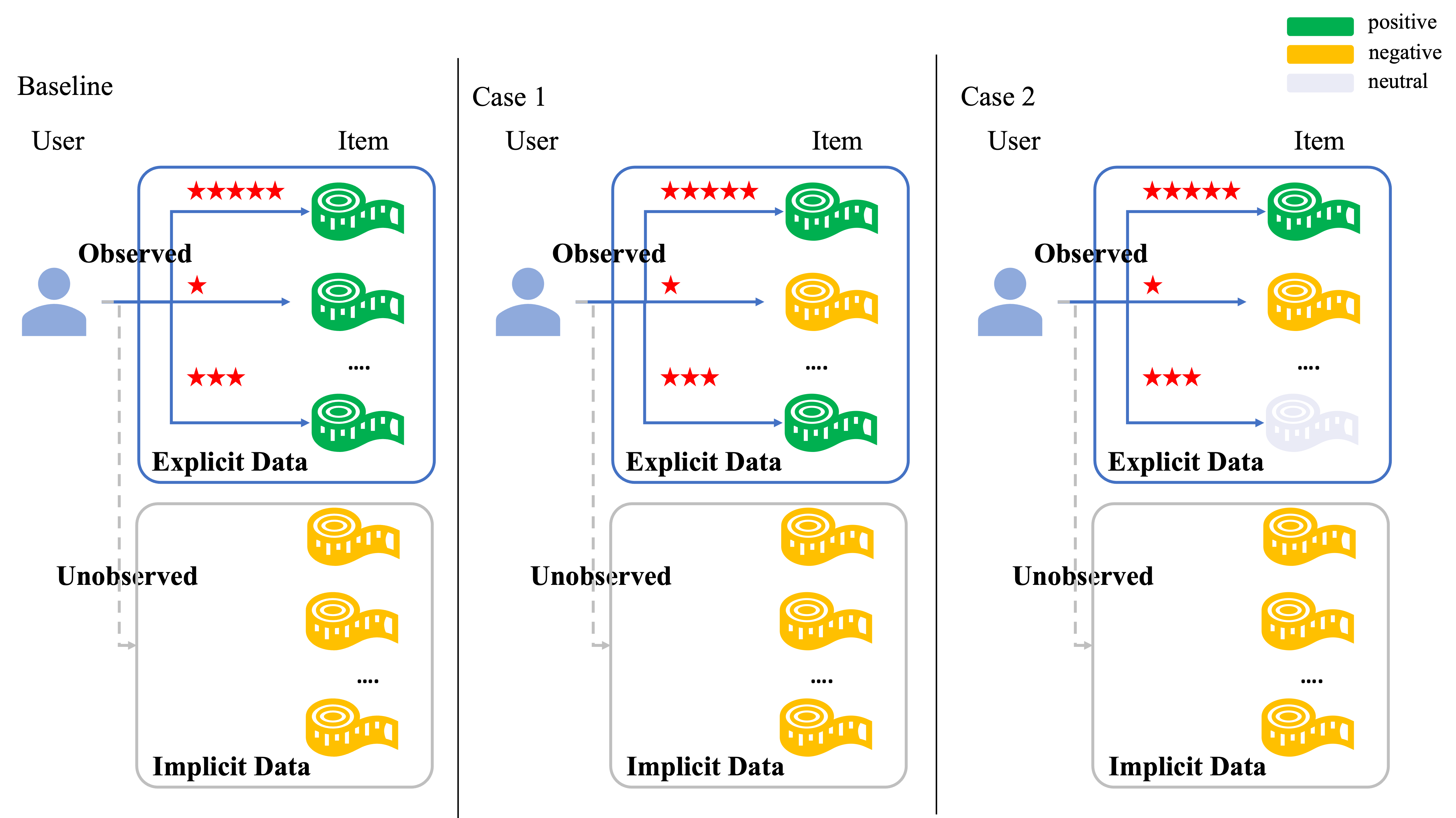
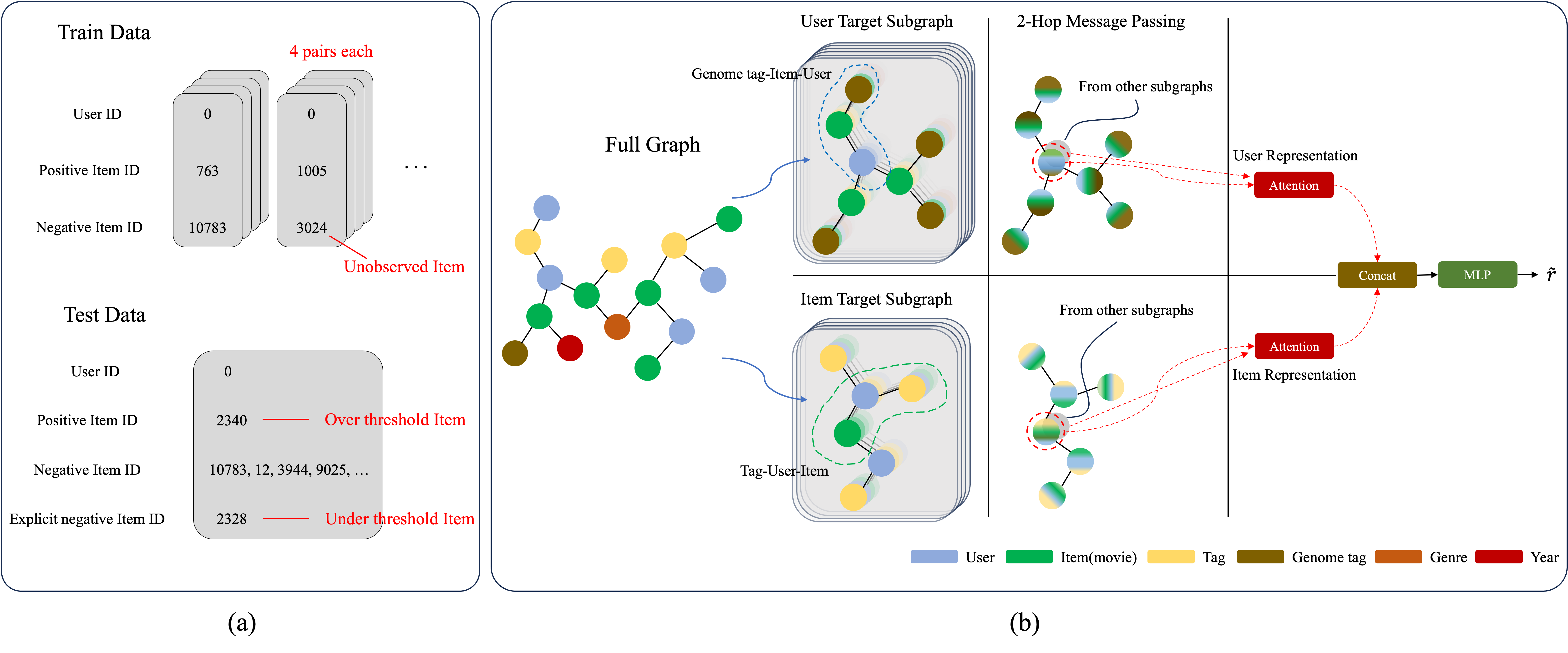
The recommendation systems based on theenhancement of the implicit and explicit sampling
Bayesian Personalized Ranking (BPR) assigns ranks to a set of items to recommend then to users. This study proposes a novel approach to improve the performance of recommendation systems. The first proposed method for the enhanced recommendation system constructs positive preferences by utilizing only the items explicitly preferred by users rather than treating all interacted items (e.g., clicked, rated, or reviewed) as positive, as was traditionally done. The second method involves using explicitly non-preferred items as negative data, and the traditional approach of using only non-interacted items as negative. Message propagation is performed on subgraphs generated through meta-path design. The results of each subgraph are used to learn the representations of users and items through an attention mechanism and graph representation learning based on this data configuration. The system then predicts scores for user-item pairs. For evaluation, the performance of the recommendation system was assessed using not only traditional accuracy metrics but also by defining a pairwise ranking accuracy metric. Pairwise ranking accuracy assigns ranks to preferred and non-preferred items to determine if the model reflects user preferences. Experimental results showed improved performance in widely used evaluation metrics for recommendation systems, such as Hit Rate and Normalized Discounted Cumulative Gain, and higher performance in pairwise ranking accuracy.
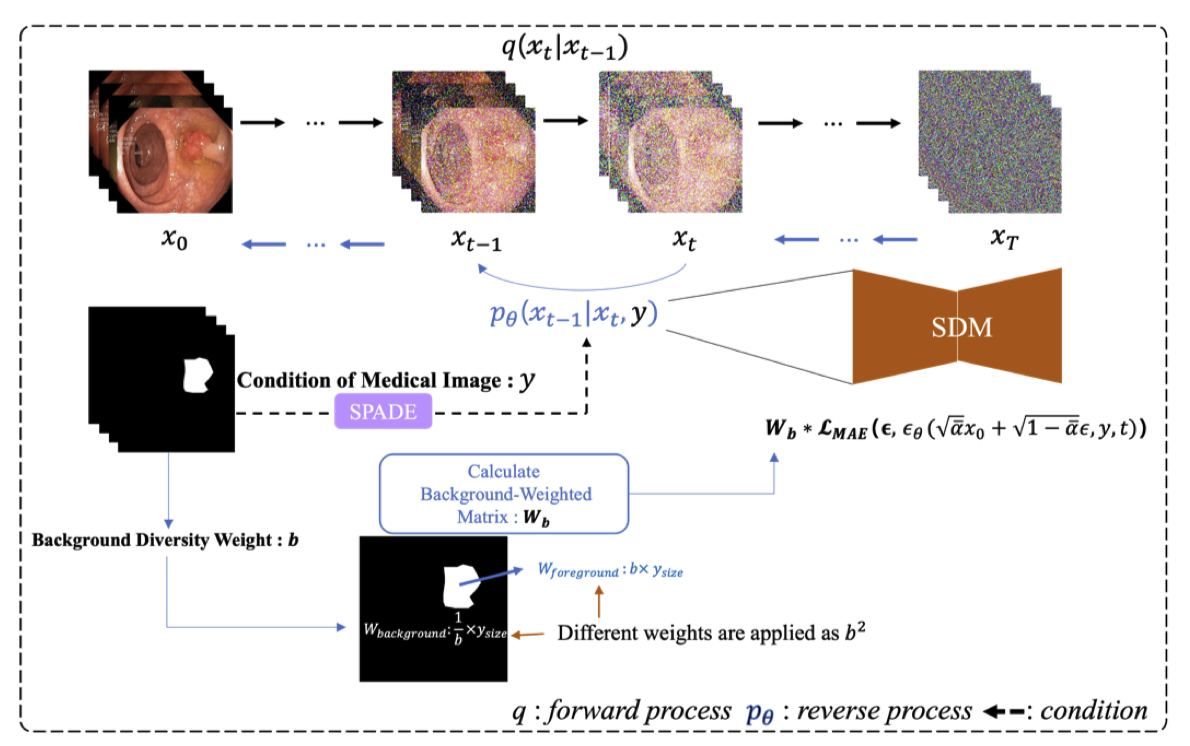
Generating Diverse Colonoscopy Images with Background-Weighted Semantic Diffusion Model
Colorectal cancer is one of the most common cancers worldwide and poses a significant threat to humans, and it is preventable through early detection of polyps. Consequently, research is actively underway to improve the performance of early detection using artificial intelligence(AI), particularl through segmentation tasks. This segmentation task requires expert annotated images of lesion areas and medical images, requiring large amounts of high-quality data. However, due to the limitations of medical data, there are constraints in obtaining high-quality data. Augmentation by image generation models has been actively researched to address this issue. In this study, we propose a model that generates diverse images by strongly applying different weights to the lesion area and the background area of the annotation, thereby improving the diverse generation capability in the background area. The evaluation involved evaluating the generated images and applying the augmented data by the model to the segmentation task to evaluate the performance improvement. Also, experimental results demonstrate the effectiveness of generating images with different backgrounds and the benefits of different image augmentations.
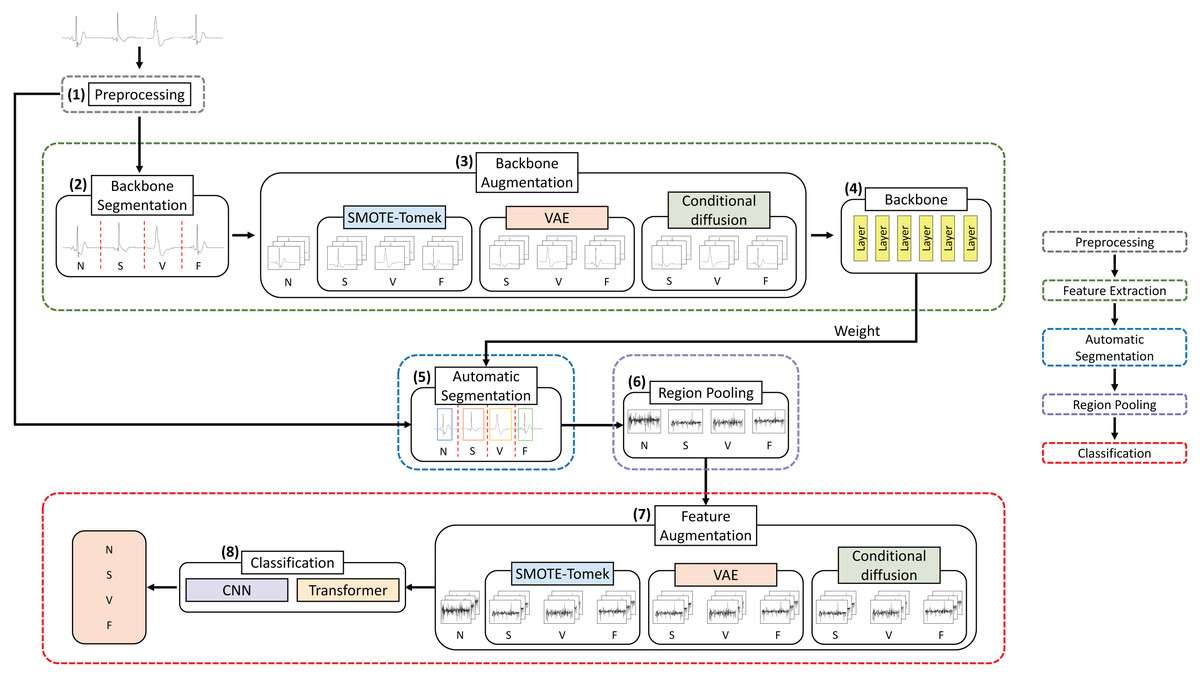
Classification of imbalanced ECGs through segmentation models and augmented by conditional diffusion model
Electrocardiograms (ECGs) provide essential data for diagnosing arrhythmias, which can potentially cause serious health complications. Early detection through continuous monitoring is crucial for timely intervention. The Massachusetts Institute of Technology-Beth Israel Hospital (MIT-BIH) arrhythmia dataset employed for arrhythmia analysis research comprises imbalanced data. It is necessary to create a robust model independent of data imbalances to classify arrhythmias accurately. To mitigate the pronounced class imbalance in the MIT-BIH arrhythmia dataset, this study employs advanced augmentation techniques, specifically variational autoencoder (VAE) and conditional diffusion, to augment the dataset. Furthermore, accurately segmenting the continuous heartbeat dataset into individual heartbeats is crucial for confidently detecting arrhythmias. This research compared a model that employed annotation-based segmentation, utilizing R-peak labels, and a model that utilized an automated segmentation method based on a deep learning model to segment heartbeats. In our experiments, the proposed model, utilizing MobileNetV2 along with annotation-based segmentation and conditional diffusion augmentation to address minority class, demonstrated a notable 1.23% improvement in the F1 score and 1.73% in the precision, compared to the model classifying arrhythmia classes with the original imbalanced dataset.

A criteria-based classification model using augmentation and contrastive learning for analyzing imbalanced statement data
Criteria Based Content Analysis (CBCA) is a forensic tool that analyzes victim statements. It involves the categorization of victims' statements into 19 distinct criteria classifications, playing a crucial role in evaluating the authenticity of testimonies by discerning whether they are rooted in genuine experiences or fabricated accounts. The exclusion of subjective opinions becomes imperative to assess statements through this forensic tool objectively. This study proposes developing an objective classification model for CBCA-based statement analysis using natural language processing techniques. Nevertheless, achieving optimal classification performance proves challenging due to imbalances in data distribution among the various criterion classifications. To enhance the accuracy and reliability of the classification model, this research employs data augmentation techniques and dual contrastive learning methods for fine-tuning the RoBERTa language model.
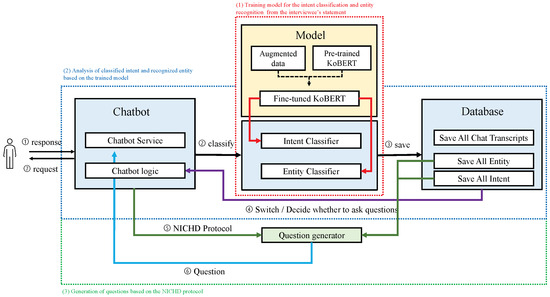
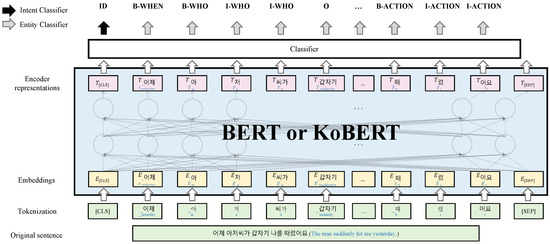
Intent Classification and Named Entity Recognition Using Augmentation
In cases of child sexual abuse, interviewing and obtaining trustworthy statements from victims and witnesses is essential because their statements are the only evidence. It is crucial to ascertain objectively the credibility of the victim’s statements, which may vary based on the nature of the questions posed by the forensic interviewer. Therefore, interview skills that eliminate subjective opinions require a high level of training for forensic interviewers. To reduce high-risk subjective interviews, objectively analyzing statements is essential. Understanding the victim’s intent and named entity recognition (NER) in the statements is necessary to give the victim open-ended questions and memory recall. Therefore, the system provides an intent classification and NER method that follows the National Institute of Child Health and Human Development Investigative Interview Protocol, which outlines the collection of objective statements. Large language models such as BERT and KoBERT, along with data augmentation techniques, were proposed using a restricted training dataset of limited size to achieve effective intent classification and NER performance. Additionally, a system that can collect objective statements with the proposed model was developed and it was confirmed that it could assist statement analysts. The verification results showed that the model achieved average F1-scores of 95.5% and 97.8% for intent classification and NER, respectively, which improved the results of the limited data by 3.4% and 3.7%, respectively.

ASL
American Sign Language (ASL) images can be used as a communication tool by determining numbers and letters using the shape of the fingers. Particularly, ASL can have an key role in communication for hearing-impaired persons and conveying information to other persons, because sign language is their only channel of expression. Representative ASL recognition methods primarily adopt images, sensors, and pose-based recognition techniques, and employ various gestures together with hand-shapes. This study briefly reviews these attempts at ASL recognition and provides an improved ASL classification model that attempts to develop a deep learning method with meta-layers. In the proposed model, the collected ASL images were clustered based on similarities in shape, and clustered group classification was first performed, followed by reclassification within the group. The experiments were conducted with various groups using different learning layers to improve the accuracy of individual image recognition. After selecting the optimized group, we proposed a meta-layered learning model with the highest recognition rate using a deep learning method of image processing. The proposed model exhibited an improved performance compared with the general classification model.
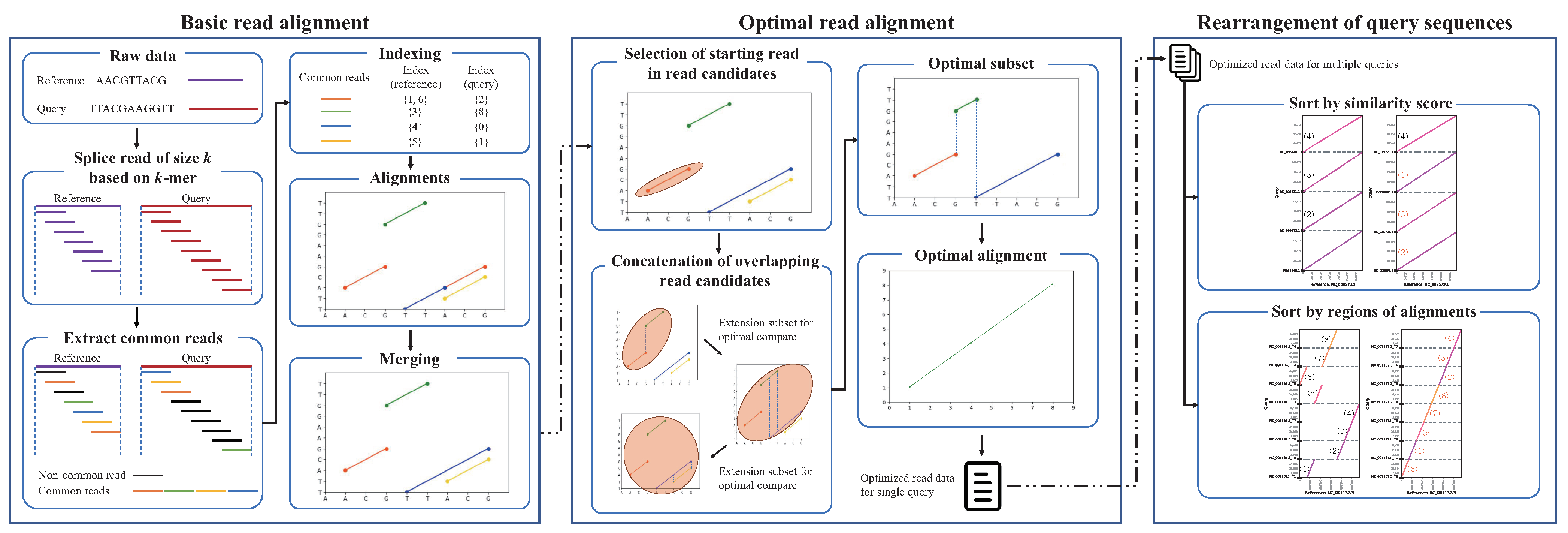
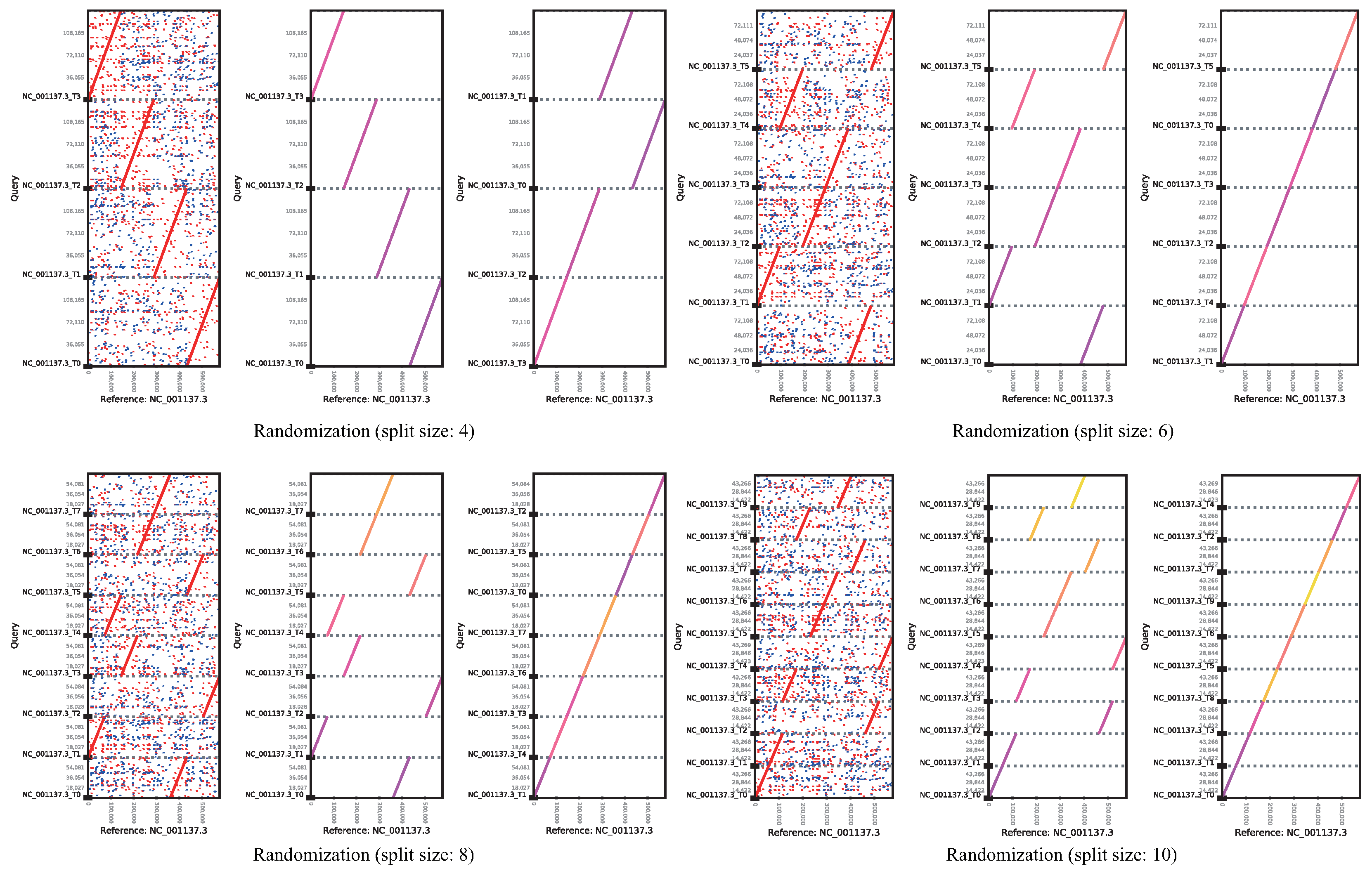
cPlot
Advances in the next-generation sequencing technology have led to a dramatic decrease in read-generation cost and an increase in read output. Reconstruction of short DNA sequence reads generated by next-generation sequencing requires a read alignment method that reconstructs a reference genome. In addition, it is essential to analyze the results of read alignments for a biologically meaningful inference. However, read alignment from vast amounts of genomic data from various organisms is challenging in that it involves repeated automatic and manual analysis steps. We, here, devised cPlot software for read alignment of nucleotide sequences, with automated read alignment and position analysis, which allows visual assessment of the analysis results by the user. cPlot compares sequence similarity of reads by performing multiple read alignments, with FASTA format files as the input. This application provides a web-based interface for the user for facile implementation, without the need for a dedicated computing environment. cPlot identifies the location and order of the sequencing reads by comparing the sequence to a genetically close reference sequence in a way that is effective for visualizing the assembly of short reads generated by NGS and rapid gene map construction.

ReGSP
The massively parallel nature of next-generation sequencing technologies has contributed to the generation of massive sequence data in the last two decades. Deciphering the meaning of each generated sequence requires multiple analysis tools, at all stages of analysis, from the reads stage all the way up to the whole-genome level. Homology-based approaches based on related reference sequences are usually the preferred option for gene and transcript prediction in newly sequenced genomes, resulting in the popularity of a variety of BLAST and BLAST-based tools. For organelle genomes, a single-reference–based gene finding tool that uses grouping parameters for BLAST results has been implemented in Genome Search Plotter (GSP). However, this tool does not accept multiple and user-customized reference sequences required for a broad homology search. Here, we present multiple-Reference–based Gene Search and Plot (ReGSP), a simple and convenient web tool that accepts multiple reference sequences for homology-based gene search. The tool incorporates cPlot, a novel dot plot tool, for illustrating nucleotide sequence similarity between the query and the reference sequences. ReGSP has an easy-to-use web interface and is freely accessible at

geneCo
In comparative and evolutionary genomics, a detailed comparison of common features between organisms is essential to evaluate genetic distance. However, identifying differences in matched and mismatched genes among multiple genomes is difficult using current comparative genomic approaches due to complicated methodologies or the generation of meager information from obtained results. This study describes a visualized software tool, geneCo (gene Comparison), for comparing genome structure and gene arrangements between various organisms. User data are aligned, gene information is recognized, and genome structures are compared based on user-defined GenBank files. Information regarding inversion, gain, loss, duplication, and gene rearrangement among multiple organisms being compared is provided by geneCo, which uses a web-based interface that users can easily access without any need to consider the computational environment.

AGORA
Next-generation sequencing (NGS) technologies have led to the accumulation of high-throughput sequence data from various organisms in biology. To apply gene annotation of organellar genomes for various organisms, more optimized tools for functional gene annotation are required. Almost all gene annotation tools are mainly focused on the chloroplast genome of land plants or the mitochondrial genome of animals. We have developed a web application AGORA for the fast, user-friendly and improved annotations of organellar genomes. Annotator for Genes of Organelle from the Reference sequence Analysis (AGORA) annotates genes based on a basic local alignment search tool (BLAST)-based homology search and clustering with selected reference sequences from the NCBI database or user-defined uploaded data. AGORA can annotate the functional genes in almost all mitochondrion and plastid genomes of eukaryotes. The gene annotation of a genome with an exon–intron structure within a gene or inverted repeat region is also available. It provides information of start and end positions of each gene, BLAST results compared with the reference sequence and visualization of gene map by OGDRAW.


Genome Search Plotter
Big data research on genomic sequence analysis has accelerated considerably with the development of next-generation sequencing. Currently, research on genomic sequencing has been conducted using various methods, ranging from the assembly of reads consisting of fragments to the annotation of genetic information using a database that contains known genome information. According to the development, most tools to analyze the new organelles’ genetic information requires different input formats such as FASTA, GeneBank (GB) and tab separated files. The various data formats should be modified to satisfy the requirements of the gene annotation system after genome assembly. In addition, the currently available tools for the analysis of organelles are usually developed only for specific organisms, thus the need for gene prediction tools, which are useful for any organism, has been increased. The proposed method—termed the genome_search_plotter—is designed for the easy analysis of genome information from the related references without any file format modification. Anyone who is interested in intracellular organelles such as the nucleus, chloroplast, and mitochondria can analyze the genetic information using the assembled contig of an unknown genome and a reference model without any modification of the data from the assembled contig.